BIP! Finder for COVID-19
Deprecation notice
The page you are trying to access is no longer available and has been deprecated. BIP4COVID19 was an open dataset and Web application offering citation-based impact indicators for COVID-19-related scientific articles, aiding literature exploration.
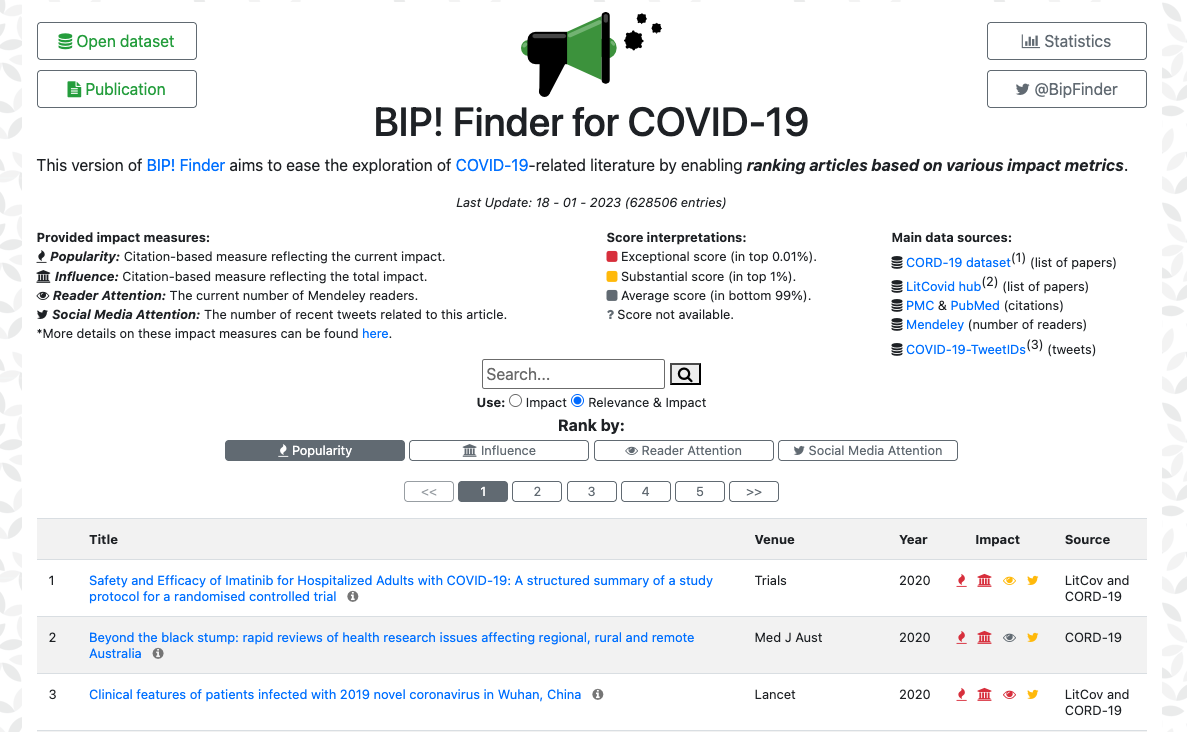
Below, you can find links to the dataset, relevant publication, as well as to the BIP! Services platform. We apologize for any inconvenience and thank you for your understanding.